Sarcomas are malignant tumors that arise from connective tissue such as bone, cartilage (the tissue lining joints), muscle, fat, tendons, and other similar tissue. These tumors may arise in any part of the body, but often occur in the head and neck, trunk, and extremities. They are often categorized according to the tissue of origin. Rhabdomyosarcoma is a tumor that arises from muscle tissue and can occur anywhere in the body, though it is most common in the head and neck region, as well as the arms and legs. The more general “soft tissue sarcoma” can arise from different types of connective tissue. These tumors are often more specifically designated “non-rhabdomyosarcoma soft tissue sarcoma” in children, though each is also named for the cell of origin, including leiomyosarcoma (from smooth muscle), liposarcoma (from fat), and fibrosarcoma (from fibrous tissue). There are many others as well. The most common in children is synovial sarcoma. The origin of this tumor is not completely known. Like rhabdomyosarcoma, these tumors can occur almost anywhere in the body. Osteosarcoma is a malignant tumor of bone. Chordomas originate in cells derived from the primitive notochord, a remnant of early development, while chondrosarcoma is a malignant tumor arising from cartilage. Ewing sarcoma is actually not a sarcoma but has traditionally been grouped in the sarcoma family due to its propensity to arise from bone and soft tissue. The most common sites are the legs and pelvic bones.
Establishing long-term tumor control for sarcoma necessitates a multidisciplinary treatment approach that entails local and systemic therapy in specific sequences validated through clinical trials. Radiation therapy is frequently used to treat local or regional disease. The sequencing of local control measures is typically derived from the tumor type, extent, and anatomic location; radiation therapy may be used postoperatively, preoperatively, or as the sole local therapy. Similar factors determine whether sarcoma patients are irradiated before, after, or with chemotherapy. Gross tumor or microscopic residual disease following surgery can often be effectively treated with local radiotherapy, as can disease that includes regional nodal involvement. Additionally, many sarcomas are infiltrative tumors with tissue beyond the resected volume harboring microscopic disease, even in the setting of apparently negative histologic margins. In these patients, preoperative radiotherapy may cause tumor shrinkage or downstaging, decrease the risk of seeding, and enhance the surgeons’ ability to obtain negative margins. Clinical trials have established the benefits of local/regional radiotherapy including limb preservation, the avoidance of disfiguring surgery, and increased local tumor control obviating a second surgery, and increased disease control that often relates to improved and improving overall survival.
Many years of experience have established that sarcomas respond best to high doses of radiation. This is particularly true for chordomas, chondrosarcomas, and osteosarcomas that arise in areas of the body that prohibit surgical removal. For example, an osteosarcoma may require over 3 times the dose of radiation required to cure a lymphoma. The delivery of radiotherapy to pediatric and young adult patients with sarcoma is uniquely complicated as the doctor must balance the appropriate dose and volume of treatment needed for local control with radiotherapy’s potential effects on developing tissues. These goals are often at odds with one another because critical normal organs, such as the spinal cord, eyes, lungs, kidneys, and salivary glands, are close to the tumor target. Childhood and adolescence is also a period of rapid bone and muscle growth. Radiation has the ability to slow or stop bone growth, depending on the age of the patient and the dose. Treatment-related effects secondary to radiation therapy, both during and after treatment, have been a major area of concern with the use of therapeutic radiation in young patients. Secondary carcinogenesis (i.e., the ability of radiation to cause a new, different tumor in the future) is observed less frequently than growth effects, but is likewise ascribed to the unique impact of ionizing radiation on developing tissues. Decades of experience have demonstrated that each of these effects (whether structural, biochemical, or functional) is related to the site of radiation treatment. The superior dose distribution of proton therapy may allow a pediatric radiation oncologist to avoid or even eliminate radiation to developing nearby normal tissues.
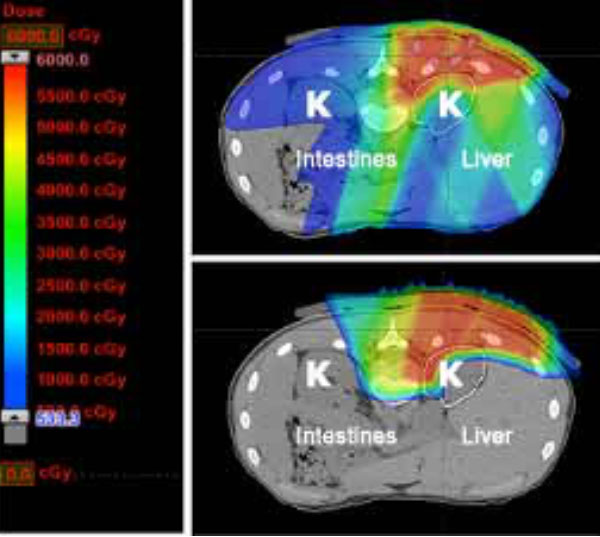
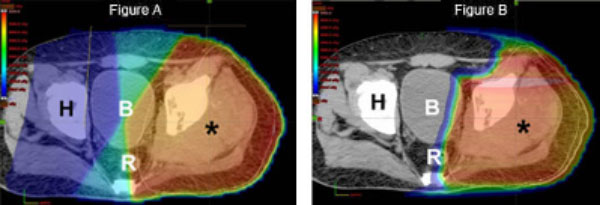
Finally, as outlined above, optimal management of sarcoma is often multimodality, meaning that both surgery and radiation are utilized to prevent a recurrence. By decreasing the radiation dose to nerves, muscle, and skin overlying the tumor site, proton therapy may decrease the chance of a surgical complication and ultimately improve a patient’s functional outcome.
In summary, radiation-related side effects for children and young adults with sarcomas treated include decrease in bone growth, soft tissue fibrosis, and muscle atrophy as well as microvascular changes which may have permanent adverse consequences. Radiation-related toxicities may be modified or enhanced by interaction with other treatment modalities, including surgery and chemotherapy. As overall survival rates continue to increase and long-term follow-up data builds, the theoretical advantage of proton therapy to reduce late effects may become even more evident in our clinics. It is likely that treatment of young sarcoma patients will yield the greatest relative benefit. Researchers at the University of Florida are international leaders in the study of proton therapy for children with sarcoma, publishing landmark articles on proton therapy for Ewing sarcoma of the pelvis1 (2020), Ewing sarcoma of the skull base2 (2020), rhabdomyosarcoma of the pelvis3 (2020), parameningeal rhabdomyosarcoma4 (2020), orbital rhabdomyosarcoma5 (2019), and chordoma6 (2021).
1Uezono H, Indelicato DJ, Rotondo RL, Mailhot Vega RB, Bradfield SM, Morris CG, Bradley JA. Treatment Outcomes After Proton Therapy for Ewing Sarcoma of the Pelvis. Int J Radiat Oncol Biol Phys. 2020 Aug 1;107(5):974-981. doi: 10.1016/j.ijrobp.2020.04.043. Epub 2020 May 8. PMID: 32437922.
2Kharod SM, Indelicato DJ, Rotondo RL, Mailhot Vega RB, Uezono H, Morris CG, Bradfield S, Sandler ES, Bradley JA. Outcomes following proton therapy for Ewing sarcoma of the cranium and skull base. Pediatric Blood Cancer. 2020 Feb;67(2):e28080. doi: 10.1002/pbc.28080. Epub 2019 Nov 17. PMID: 31736243.
3Indelicato DJ, Rotondo RL, Krasin MJ, Mailhot Vega RB, Uezono H, Bradfield S, Agarwal V, Morris CG, Bradley JA. Outcomes Following Proton Therapy for Group III Pelvic Rhabdomyosarcoma. Int J Radiat Oncol Biol Phys. 2020 Apr 1;106(5):968-976. doi: 10.1016/j.ijrobp.2019.12.036. Epub 2020 Jan 25. PMID: 31987977.
4Bradley JA, Indelicato DJ, Uezono H, Morris CG, Sandler E, de Soto H, Mailhot Vega RB, Rotondo R. Patterns of Failure in Parameningeal Alveolar Rhabdomyosarcoma. Int J Radiat Oncol Biol Phys. 2020 Jun 1;107(2):325-333. doi: 10.1016/j.ijrobp.2020.01.035. Epub 2020 Feb 7. PMID: 32044412.
5Indelicato DJ, Rotondo RL, Mailhot Vega RB, Uezono H, Bradfield S, Agarwal V, Hol ML, Bradley JA. 45 GyRBE for group III orbital embryonal rhabdomyosarcoma. Acta Oncol. 2019 Oct;58(10):1404-1409. doi: 10.1080/0284186X.2019.1627412. Epub 2019 Sep 18. PMID: 31530120.
6Indelicato DJ, Rotondo RL, Mailhot Vega RB, Holtzman AL, Looi WS, Morris CG, Sandler ES, Aldana PR, Bradley JA. Local Control After Proton Therapy for Pediatric Chordoma. Int J Radiat Oncol Biol Phys. 2021 Apr 1;109(5):1406-1413. doi: 10.1016/j.ijrobp.2020.11.051. Epub 2020 Nov 27. PMID: 33253819.
Engaging young patients

Survivor Spotlight

Types of pediatric cancer treated with proton therapy include: